Supervisors


Prof. Pradeepa Bandaranayake
Authors

S.Thinesh

K.Sathursan

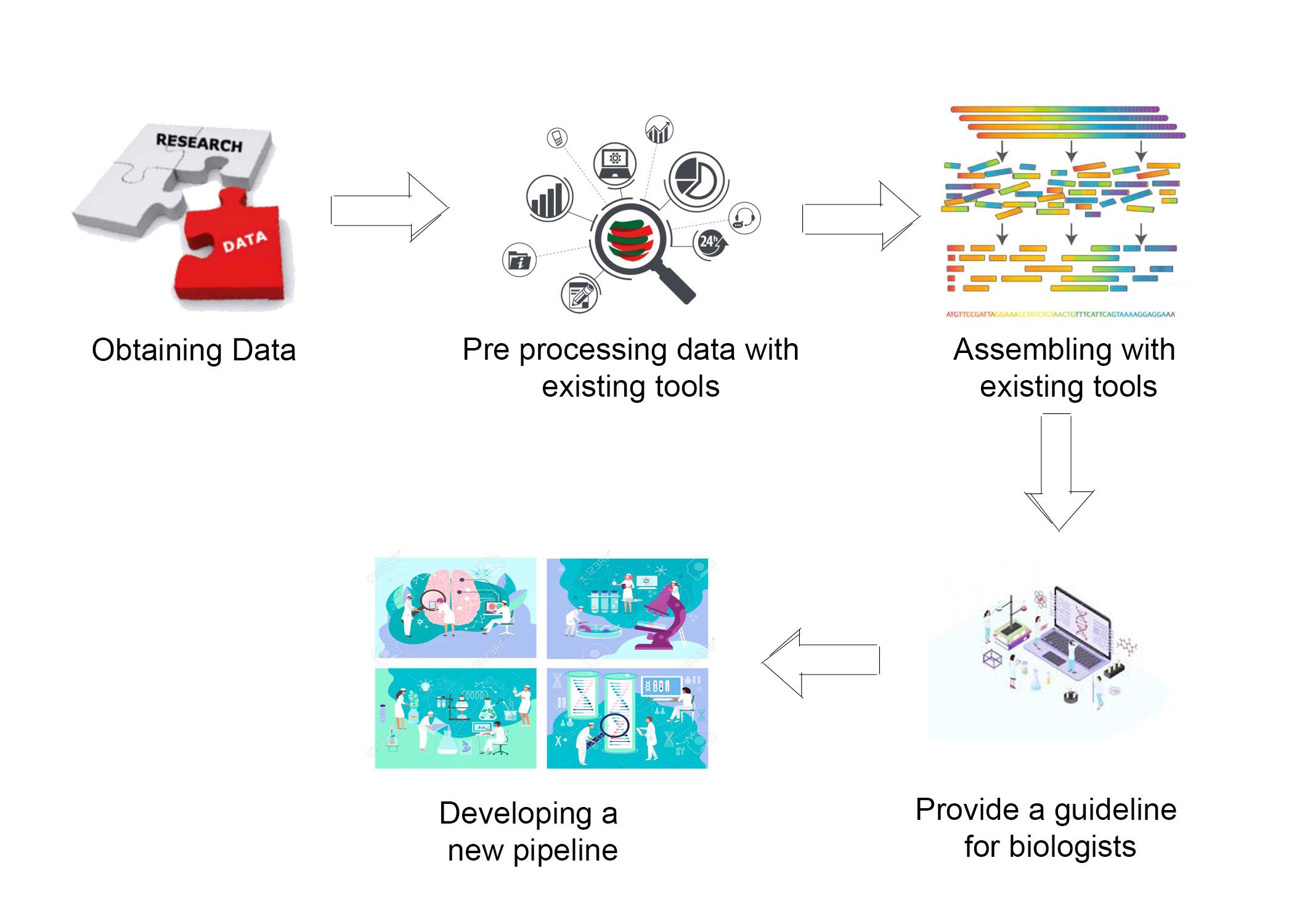
Genome sequencing and genome assembly are the computational process of converting the sequence composition of the gene within the cell of an organism in a human readable form. Mitochondria is an important genome in the cell and there is a need to study this genome for various reasons. The process of determining the order of bases A,G,T,C in the genome is known as genome sequencing.While sequencing the genome the original genome is separated into huge number of small parts known as reads and the end results of sequencing is a huge pool of reads(strings of A,G,T,C). These reads must be assembled back in a computer so that a biologist can identify and annotate the functionality of it. There are several techniques were developed to sequence the genome, the modern approach is next generation sequencing. Ilumina sequencing is one of the broadly used next generation sequencing method and this method produce large number of high precision sequencing short reads whereas other older methods produce longer reads.So the computational complexity of assembling back this large amount of read is high but cost efficient. Here we mainly discuss on low coverage sequencing data assembly.The sequencing data consist of multiple copies of same genome in low coverage data the number of copies are relatively lower than high coverage data.
In this project we examined the tools used for mitochondrial genome assembly by assembling different datasets and measured the parameters that make impact in the assembly process. From the results we obtained from the experiment we made decisions of doing mitochondrial genome assembly.